





Abstract
The biological mechanisms of autism spectrum disorders (ASDs) are largely unknown in spite of extensive research. ASD is characterized by altered function of multiple brain areas including the temporal cortex and by an increased synaptic excitation:inhibition ratio. While numerous studies searched for evidence of increased excitation in ASD, fewer have investigated the possibility of reduced inhibition. We characterized the cortical γ-amino butyric acid (GABA)ergic system in the rat temporal cortex of an ASD model [offspring of mothers prenatally injected with valproic acid (VPA)], by monitoring inhibitory post-synaptic currents (IPSCs) with patch-clamp. We found that numerous features of inhibition were severely altered in VPA animals compared to controls. Among them were the frequency of miniature IPSCs, the rise time and decay time of electrically-evoked IPSCs, the slope and saturation of their input/output curves, as well as their modulation by adrenergic and muscarinic agonists and by the synaptic GABAA receptor allosteric modulator zolpidem (but not by the extra-synaptic modulator gaboxadol). Our data suggest that both pre- and post-synaptic, but not extra-synaptic, inhibitory transmission is impaired in the offspring of VPA-injected mothers. We speculate that impairment in the GABAergic system critically contributes to an increase in the ratio between synaptic excitation and inhibition, which in genetically predisposed individuals may alter cortical circuits responsible for emotional, communication and social impairments at the core of ASD.
Introduction
Autism spectrum disorders (ASDs) are a group of developmental conditions defined by the convergence of core symptoms consisting of repetitive behaviour and limited range of interests, impaired communication and decreased social interaction (DSM-IV). ASD is often accompanied by hyperexcitable states, including aggression (Parikh et al. 2008), anxiety (Spiker et al. 2012), hyperactivity (Antshel et al. 2011), epilepsy (Tuchman et al. 2010; Tuchman and Cuccaro, 2011), as well as a plethora of sleep disorders (Goldman et al. 2009; Souders et al. 2009; Thatcher et al. 2009; Glickman, 2010). The reliable presence of cell–cell communication abnormalities in such heterogeneous group of conditions, as in fragile X (Selby et al. 2007; Bassell and Warren, 2008), Rett (Calfa et al. 2011) and Angelman syndromes (Roden et al. 2010) and the tuberous sclerosis complex (Curatolo et al. 2008) suggests that impaired synaptic function is an important factor in the aetiology and general mechanisms of ASDs. Together, these data led to the hypothesis that an increase in the ratio between synaptic excitation and inhibition during a critical period might determine a dysfunctional development of the neural circuits causing the core symptoms of ASD (Rubenstein and Merzenich, 2003; Gogolla et al. 2009).
An increased synaptic excitation:inhibition ratio can be caused by increased glutamatergic signalling, decreased synaptic inhibition, or a combination of the two. The use of an environmental animal model of autism based on the offspring of mother rats injected with valproic acid (VPA; Schneider and Przewlocki, 2005; Schneider et al. 2007, 2008), which epidemiologic data indicate as an important environmental cause for ASD (Moore et al. 2000; Williams et al. 2001; Rasalam et al. 2005), suggests that the number of excitatory synapses in local cortical circuits is abnormally enhanced in ASD (Rinaldi et al. 2008a, b). We used the same animal model to test the possibility of impaired efficacy of γ-amino butyric acid (GABA)ergic synaptic function, which is under increasing scrutiny as a potentially critical factor in the development of ASDs (Hussman, 2001; Pizzarelli and Cherubini, 2011).
We found that pre-synaptic and post-synaptic, but not extra-synaptic, GABAergic function and its modulation are severely compromised in VPA animals compared to controls. These data raise the possibility that a derangement in the developing GABAergic system increases the ratio between synaptic excitation and inhibition, bringing about a critical contribution to core symptoms of ASD.
Method
Animals
Sprague–Dawley female rats (Charles River, USA) were mated overnight with Sprague–Dawley males and pregnancy was determined by the presence of a vaginal plug (first day of gestation). Pregnant mother rats were given a single i.p. injection of VPA (600 mg/kg in 0.9% saline) on day 12.5 of pregnancy and control rats were injected with the same volume of saline alone as described previously (Schneider and Przewlocki, 2005). The offspring were weaned on post-natal day (PD) 21 and rats of either sex were housed separately. All experimental procedures comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University committee on Animal Research at the University of Texas at Dallas. Offspring from PD 23–45 were used for electrophysiology experiments. We used a temporal cortex slice preparation as described in the Supplementary Material.
Preparation
We used a temporal cortex slice preparation similar to the one previously described (Atzori et al. 2001). Animals were anaesthetized with isoflurane (Baxter, USA) and killed according to the National Institutes of Health Guidelines (UTD IACUC number 04-04). Their brains were sliced with a vibratome (VT1000; Leica, USA) in a cold solution (0–4 °C) containing (mm) 126 NaCl, 3.5 KCl, 10 glucose, 25 NaHCO3, 1.25 NaH2PO4, 1.5 CaCl2, 1.5 mgCl2 and 0.2 ascorbic acid, at pH 7.4 and saturated with a mixture of 95% O2 and 5% CO2 (ACSF). Coronal slices (270 µm thick) from the most caudal fourth of the brain were retained after removing the occipital convexity (caudal end of the brain after removal of the cerebellum) and subsequently incubated in ACSF at 32 °C before being placed in the recording chamber. The recording area was selected dorsally to the rhinal fissure corresponding to the auditory cortex (Rutkowski et al. 2003). The recording solution used to detect the miniature inhibitory post-synaptic currents (mIPSCs) contained tetrodotoxin (0.5 µm). The recording solution also contained 6,7-dinitroquinoxaline-2,3-dione (10 µm) and kynurenate (2 mm) for blocking α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor and N-methyl-d-aspartate receptor-mediated currents, respectively.
Drugs
All drugs were purchased from Sigma (USA), Tocris (USA) and Alomone Laboratories (Israel). Stock solutions of all drugs were prepared in water except for oxotremorine, of which stock solution (10 mm) was prepared in ethanol. For non-aqueous solutions, the final concentration of the solvent was added to the recording control solution. After recording an initial baseline for 7–10 min, drugs were bath-applied for 5 min or longer, until reaching a stable condition.
Electrophysiology
Slices were placed in an immersion chamber, where cortical layer II/III cells with a prominent apical dendrite, suggestive of pyramidal morphology, were visually selected using a BX51 microscope (Olympus, USA) with an infrared camera system (DAGE-MTI, USA). IPSCs were recorded in the whole-cell configuration, in voltage-clamp mode, holding the membrane potential at a holding voltage Vh = − 60 mV, with 3–6 MΩ electrodes filled with a solution containing (mm): 100 CsCl, 5 1,2-bis(2-aminophenoxy) ethane-N,N,N,N-tetraacetic acid K, 1 lidocaine N-ethyl bromide, 1 mgCl2, 10 Hepes, 4 glutathione, 1.5 ATPMg2, 0.3 GTPNa2 and 20 phosphocreatine. The intra-cellular recording solution was titrated at pH 7.3 and had an osmolarity of 270 mOsm. The holding voltage was not corrected for the junction potential (<4 mV). Electrically-evoked IPSCs (eIPSCs) were measured by delivering two electric stimuli (90–180 µs, 10–50 µA) 50 ms apart every 20 s with an isolation unit, through a glass stimulation using monopolar electrode filled with ACSF and placed 150–200 µm away from the recording electrode. A 2-mV voltage step was applied at the beginning of every episode to monitor the quality of the recording. Recordings where series resistance changed by >20% were discarded from the analysis. In the analysis of miniature post-synaptic currents, only single events were considered for kinetic analysis. Miniature events were analysed with the Clampfit software (Axon, USA). The average number of events per each condition was >300.
Statistical analysis
We defined a statistically stable period as a time interval (3–5 min) along which the IPSC mean amplitude measured during any 1-min assessment did not vary according to a paired Student's t test. Means±s.e.m. are reported. Paired pulse ratio (PPR) was calculated as the mean of the second response divided by the mean of the first response, according to Kim and Alger (2001). Drug effects were assessed by measuring and comparing the different parameters (IPSC mean amplitude, PPR and others) between controls vs. VPA animals, with paired Student's t test. Data were reported as different only if p < 0.05% (* p < 0.05, ** p < 0.02), unless indicated otherwise. Quantal analysis was performed to identify the locus of defect in VPA animals affecting post-synaptic current amplitudes. We used a coefficient of variation (CV) analysis similar to the one reported by Faber and Korn (1991) and by Zucker and Regehr (2002) to determine the synaptic locus.
Results
The temporal lobes of mammals including rats, humans and other species host multiple auditory and communication-related areas and are critically impaired in ASD (Hazlett et al. 2011; Suzuki et al. 2011). For this reason we compared the temporal cortex in VPA vs. control animals using electrophysiological recording.
Miniature IPSC impairment in VPA animals
We compared the mIPSCs, recorded in the presence of the Na+ channel blocker tetrodotoxin (0.5 µm) between control and VPA animals. Representative traces are shown in Fig. 1a. The frequency of the mIPSC was significantly reduced in VPA animals compared to controls (1.39 ± 0.21 Hz in VPA vs. 3.05 ± 0.64 Hz in controls, n = 10, p < 0.05, Student's t test; Fig. 1b), while the mean mIPSC amplitude did not differ significantly (Fig. 1c). Analysis of mIPSC kinetics revealed that both rise and decay time were significantly slower in the VPA animals (rise time: 0.81 ± 0.05 ms in VPA vs. 0.63 ± 0.03 ms in controls, n = 8; p < 0.05, unpaired Student's t test; decay time: 33.0 ± 5.7 ms in VPA vs. 18.1 ± 3.3 ms in controls, p < 0.02, unpaired Student's t test, n = 10; Fig. 1f). While changes in mIPSC frequency suggest pre-synaptic differences, different kinetics would indicate post-synaptic alterations in GABAergic transmission.
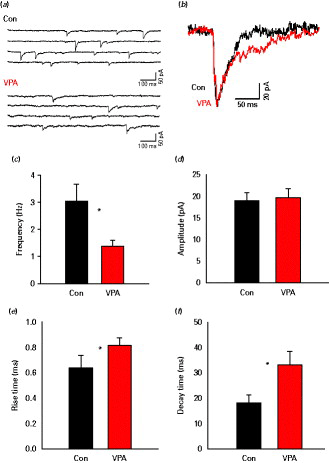
Miniature inhibitory post-synaptic current (mIPSCs) in control and valproic acid (VPA) animals. (a) Representative traces of mIPSCs from layer II/III pyramidal cells of control and VPA animals in the presence of tetrodotoxin (0.5 µm). (b) Average of 20 aligned mIPSCs in control or VPA animals (expanded scaling; control, black; VPA, grey). (c and d) Average frequency and amplitude, respectively, of mIPSCs in control and VPA (n = 10 each). (e and f) The mIPSC rise time and decay time in VPA and controls. VPA mIPSC displayed a lower frequency and slower kinetics compared to control animals. * p < 0.05.
Input/output response of electrically-evoked IPSCs
While the source of mIPSCs is not exactly known, electrically-evoked synaptic currents likely originate from stimulation of axons surrounding the stimulation electrode. We obtained eIPSCs by stimulating cortical layer II/III at about 200 µm from the recorded cell. We compared GABAergic transmission between control and VPA animals and determined the synaptic output as a function of increasing stimulation intensity (see Method). Each point in the input/output (I/O) curves corresponds to averaged responses over 4–10 extracellular electrical responses delivered at the same intensity (example of I/O curve in Fig. 2a). For each recording three parameters were extracted: response threshold; initial slope; saturation current. The threshold was the smallest intensity eliciting a non-zero synaptic response. The initial slope was calculated between the first two non-null responses of each curve. The saturation current was the maximum eIPSC amplitude observed and typically did not significantly change in the rightmost part of the I/O curve. While the response threshold (Fig. 2b) was similar in control (30 ± 5 µA, n = 12) and VPA animals (20 ± 10 µA, n = 10, n.s., unpaired Student's t test), eIPSC slope and saturation levels were significantly lower in VPA animals compared to controls (slope 1.4 ± 0.4 pA/µA in VPA, 0.35 ± 0.15 in control, p < 0.02; saturation 165 ± 42 pA in VPA, vs. 841 ± 148 pA in control, p < 0.02; Fig. 2c, d) indicating that GABAergic transmission in VPA animals did not even reach 20% of control.
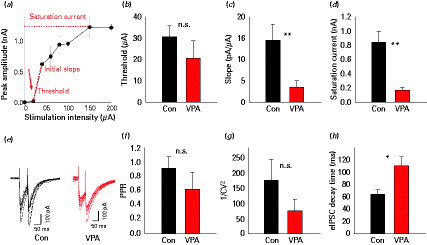
Basal inhibitory synaptic transmission in control and valproic acid (VPA) animals. (a) Example of input/output (I/O) curve. Each trace corresponds to the averaged responses over at least four extracellular electrical pulses delivered at the same intensity. (b–d) I/O curve threshold, slope, and saturation level, respectively, in control and VPA animals. (e) Representative traces of 10 evoked inhibitory post-synaptic current (eIPSCs) in control (black) and VPA (grey) animals. (f) Paired pulse ratio (PPR) A2/A1 in control and VPA animals. (g) Coefficient of variation (1/CV2) analysis, averages for control (black) and VPA (grey). (h) Decay time in control (black) and VPA (grey) animals. * p < 0.05, ** p < 0.02.
Analysis of paired pulse stimulation at an interpulse delay of 50 ms (example in Fig. 2e, f) showed that PPR and CV did not significantly differ between VPA and control animals (representative traces in Fig. 2e, f for control and VPA respectively; PPR 1.02 ± 0.04 in control vs. 0.90 ± 0.12 in VPA, p > 0.05; 1/CV2 175 ± 71 in control vs. 77 ± 36 in VPA, n.s.). Consistent with the finding of the mIPSC, the decay time was slower in VPA animals compared with controls (τ = 64.3 ± 6.3 ms in control vs. 110.3 ± 15.4 ms in VPA animals, p < 0.05). These results indicate the presence of a deficit in the GABAergic transmission of VPA animals.
Synaptic but not extra-synaptic GABAAR-mediated currents are selectively impaired in VPA animals
GABAA receptors (GABAARs) in the central nervous system are localized both synaptically as well as extra-synaptically (Drasbek and Jensen, 2006). In order to determine the contribution of synaptic GABAARs we recorded layer II/III eIPSC response to zolpidem, an allosteric modulator of GABAARs that preferentially binds to α1 and α2/3 subunits of GABAARs located mostly within post-synaptic densities (Jacob et al. 2008). While we found that eIPSC in both VPA and control animals were enhanced after application of zolpidem (0.5 µm), the extent of zolpidem-induced increase in eIPSC amplitude was significantly smaller in VPA animals (+29 ± 4%, n = 9), compared to control animals (+55 ± 1.7%, p > 0.05, n = 12, unpaired Student's t test). Figure 3a, b shows representative examples of the eIPSC amplitude time-course following zolpidem application in control and VPA animals respectively. The mean increment is shown in Fig. 3c.
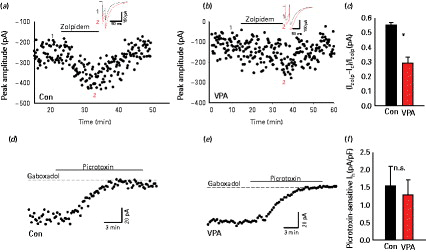
Different modulation of synaptic and extra-synaptic GABAA receptors (GABAAR)-mediated currents: (a) Bath-application of zolpidem (500 nm) showing a decrease of eIPSC amplitude in control animals. The insert shows an example of an evoked inhibitory post-synaptic current (eIPSC) at an expanded scaling control (black) zolpidem (grey). The numbers 1 and 2 in the insert refer to the average of four traces in the amplitude time-course. (b) Same as (a) but in valproic acid (VPA) animals; time-course and traces (insert) showing the effect of zolpidem (500 nm) in eIPSC; control (black), zolpidem (grey). (c) Shows normalized zolpidem-induced change of eIPSC amplitude in control and VPA animals (n = 9). (d and e) Change in GABAAR-mediated tonic currents (in 10 µm 6,7-dinitroquinoxaline-2,3-dione and 2 mm kynurenic acid). Examples of time-course of the bath-application of gaboxadol (5 µm), a specific agonist for the extrasynaptic δ subunit of GABAAR, which caused an increase of tonic conductance, revealed by increased holding current blocked by picrotoxin (100 µm), in control and VPA animals, respectively. (f) Mean of picrotoxin sensitive current in gaboxadol normalized to the cell capacitance, control and VPA animals (n = 11). * p < 0.05.
Extra-synaptic currents are an important determinant of neuronal excitability. 4,5,6,7-Tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP, gaboxadol) binds specifically to δ subunits present extra-synaptically (Drasbek and Jensen, 2006). We estimated the response of extra-synaptic GABAAR by determining the change in picrotoxin sensitive holding current following bath application of gaboxadol (5 µm). Figure 3d, e display representative time-course of the tonic current from layer II/III neurons in control and VPA animals respectively, measured as picrotoxin-sensitive component of the holding current following application of gaboxadol (Drasbek and Jensen, 2006). The tonic current did not significantly differ between control and VPA animals (37 ± 4.8% pA, n = 10 vs. 30 ± 7.6% pA in control, n = 9, n.s., Student's t test; Fig. 3f). Altogether, these results suggest a selective impairment of GABAARs.
Impaired pre-synaptic modulation of inhibitory transmission in VPA animals
A lower frequency in mIPSCs raises the possibility of pre-synaptic impairment in VPA animals. Previous studies from our laboratory have shown that GABAergic terminals are subject to intense pre-synaptic modulation by adrenergic and muscarinic receptors. In fact the activation of pre-synaptic adrenoceptors increases eIPSC amplitude (Salgado et al. 2011), while activation of muscarinic receptors decreases eIPSC amplitude (Salgado et al. 2007). We considered the possibility that GABAergic terminals in VPA animals responded to adrenergic or muscarinic agonists differently from controls. As in the previous series of experiments, eIPSCs were recorded from cortical layer II/III neurons after stimulation of the same layer.
As expected, bath application of norepinephrine (NE) increased the eIPSC amplitude from 405 ± 27 pA to 630.7 ± 41 pA in control animals, corresponding to 68.5 ± 7.1% (see example of eIPSC amplitude time-course in Fig. 4a). On the contrary, NE failed to show a similar increase in eIPSC modulation in VPA animals from 85.5 ± 15.25 pA to 103.23 ± 10.8 pA after NE, corresponding to 25 ± 6% increase (n = 12, p < 0.05; Fig. 4a). The mean eIPSC amplitudes are shown for control and VPA animals in Fig. 4b (n = 12, p < 0.05%; n = 11, n.s.). Also, while in control animals NE significantly decreased PPR, NE failed to affect PPR in VPA animals (PPR = 1.08 ± 0.17 at baseline vs. 1.01 ± 0.15 after NE, n = 12 n.s., compared to 1.13 ± 0.03 at baseline vs. 0.86 ± 0.04 in NE in control animals; Fig. 4c).
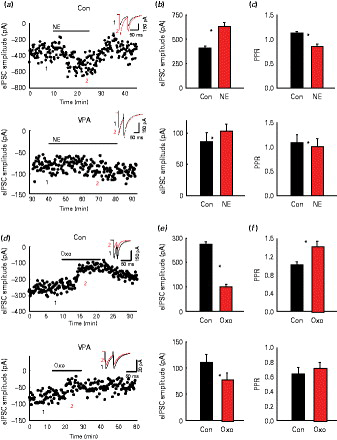
Differential norepinephrine (NE) and oxotremorine (Oxo) on modulation of evoked inhibitory post-synaptic currents (eIPSCs): (a) Time-course and traces (in the inserts) showing the effect of bath application of NE (20 µm) on eIPSC amplitude in control and valproic acid (VPA) animals, respectively (insert: baseline black; NE grey). The numbers 1 and 2 in the insert refer to the average of four traces in the amplitude time-course. (b) Average NE-induced change in eIPSC amplitude in control and VPA animals, respectively. (c) Average NE-induced change in paired pulse ratio (PPR) in control and VPA animals, respectively (d) Time-course and traces (in the inserts) showing the effect of bath application of Oxo (10 µm) on eIPSC amplitude in control and VPA animals, respectively (insert: baseline, black, Oxo, grey). (e) Average Oxo-induced change in eIPSC amplitude in control and VPA animals, respectively. (f) Average Oxo-induced change in PPR in control and VPA animals, respectively. * p < 0.05.
We also compared the effects of the muscarinic agonist oxotremorine on eIPSC amplitude and PPR. We found that oxotremorine decreased eIPSC amplitude both in control (from 275 ± 10 pA to 102 ± 9 pA, n = 12, p < 0.02; example of eIPSC amplitude time-course in Fig. 4d) and VPA animals (from 110.8 ± 15.1 pA to 78.9 ± 11.21 pA, n = 9, p > 0.05; example of eIPSC amplitude time-course in Fig. 4d). However, the extent of the oxotremorine-induced decrease in eIPSC amplitude significantly differed between control and VPA animals (68 ± 5% in control animals vs. 28 ± 5% in VPA animals, p < 0.05). Mean eIPSC amplitudes are shown in Fig. 4e for control and VPA animals, respectively. PPR changed significantly in control rats (1.0 ± 0.07 in baseline vs. 1.43 ± 0.10 in oxotremorine, n = 17, p < 0.05%; Fig. 4f) whereas it did not change in VPA animals (PPR = 0.63 ± 0.09 in baseline vs. 0.72 ± 0.07 in oxotremorine, n = 12, n.s.). Altogether, these data suggest that pre-synaptic regulation of GABAergic terminals is quantitatively and qualitatively altered in VPA animals.
Discussion
Our results show for the first time that GABAergic transmission is severely compromised in VPA animals. While the decreased frequency of mIPSCs qualitatively resembles the data found in the Rett syndrome model (Zhang et al. 2008), suggestive of a pre-synaptic deficit, the slower kinetic, typical of immature neurons (Cohen et al. 2000) indicated a post-synaptic alteration.
Post-synaptic impairment of GABAergic transmission in VPA synapses
The remarkable decrease in slope and saturation level (down to only ∼20% of control) in I/O curves and the absence of significant changes in PPR or CV further supported a dramatic impairment of post-synaptic function in VPA animals. The reduced sensitivity of VPA animals to zolpidem, which enhances GABAergic currents by preferentially binding to intra-synaptic GABAAR α subunits, suggested that VPA inhibitory synapses might have a different subunit composition compared to controls, in agreement also with the different kinetics of mIPSCs. A different sensitivity to zolpidem of GABAAR-mediated currents was also found in oocytes injected with GABAARs from Angelman patients (Roden et al. 2010). The low sensitivity of VPA inhibitory synapses to zolpidem could be due to the insertion in the membrane of GABAARs from extra-synaptic or even intra-cellular origin. Different from synaptic GABAergic currents, we found no changes in the extent of tonic GABAergic currents, measured as the picrotoxin-sensitive component of the holding current after application of THIP, a specific allosteric agonist of extra-synaptic GABAARs (Drasbek and Jensen, 2006). A different response to zolpidem, but not to THIP, could be due to an abnormal delivery of synaptic-specific subunits of GABAARs to GABAergic synapses in VPA animals, consistent with a failure of normal endosome-membrane trafficking of GABAAR subunits postulated in ASD (Momoi et al. 2010). These data are also consistent with one of the earliest reports of a decrease in GABAAR density, in contrast to a normal distribution of glutamatergic, serotoninergic and cholinergic receptors, obtained using neurochemical techniques (Blatt et al. 2001), as well as with a decreased density of benzodiazepine binding sites (Oblak et al. 2009, 2011) and GABAAR (Fatemi et al. 2009; Fukuchi et al. 2009) in autoptic brains from ASD sufferers.
Also, in line with our results is the observation that some subtypes of ASDs are linked to anomalies in chromosomal region 15q11-q13, which possesses a gene cluster encoding for multiple subunits of the GABAAR (Shao et al. 2003). Interestingly, deletion of the gene encoding for the β3 subunit of the GABAAR in mouse displays epilepsy, as well as symptoms resembling Angelman syndrome, another disorder of the autistic spectrum (Homanics et al. 1997; DeLorey et al. 1998, 2008).
Deficit in pre-synaptic modulation
The presence of post-synaptic alterations does not exclude the possibility of concomitant pre-synaptic changes. GABAergic axons in autoptic brains from ASD patients display marked decrease in the levels of glutamate decarboxylase (Fatemi et al. 2002; Yip et al. 2007, 2009; Buttenschon et al. 2009), suggesting an impairment of interneuronal pre-synaptic function. In our experiments, the decreased mIPSC frequency, the failure of GABAergic axons to display sensitivity to NE (>60% increase in controls but only ∼10% in VPA) and the milder deficit in muscarinic modulation (∼67% in controls but only ∼27% in VPA), might be among multiple impairments in local interneuronal pre-synaptic function.
Alterations in cortical interneuronal circuitry might be caused by failure of proper differentiation, migration and/or proliferation of GABAergic neurons (Schmitz et al. 2005), which control the balance between synaptic excitation and inhibition (Freund, 2003). Further support to the hypothesis of an impaired inhibitory system in ASD comes from changes in the distribution of GABAergic neurons in autoptic tissue from autistic patients (Lawrence et al. 2010) and fragile X syndrome patients (Selby et al. 2007), paralleled by altered expression and density of GABAergic markers in other brain areas in VPA (Dendrinos et al. 2011) and other animal models of ASD (Gogolla et al. 2009), detected for parvalbumin-, calbindin- and calretinin-positive interneurons (Lawrence et al. 2010). The slower mIPSC kinetics might be due to a selective decrease in number or effectiveness of somatic inhibitory synapses between interneurons and pyramidal cells, possibly caused by an early dysfunction in chemo-attractive/repulsive mechanisms regulating the proper development of immature GABAergic cells (Flames et al. 2004; Hernandez-Miranda et al. 2010).
Functional consequences of the GABAergic deficit
Numerous earlier studies have reported an enhanced excitatory transmission and long-term plasticity in VPA animals (Rinaldi et al. 2007; Silva et al. 2009). These and other work suggested the ‘intense world theory’ of ASD (Markram et al. 2007; Markram and Markram, 2010), in which an abnormally elevated cortical local excitability would give rise to emotional and sensory hypersensitivity characteristic of ASD patients. An impaired GABAergic system would produce an additional shift in the excitatory:inhibitory ratio, further contributing to local hyperexcitability. Altogether, these data converge to support the hypothesis that in spite of the heterogeneity in the range of genetic, epigenetic and environmental factors at the root of ASD, an increased ratio between cortical synaptic excitation and inhibition might be a shared mechanism in ASD (Rubenstein and Merzenich, 2003; Yizhar et al. 2011). This hypothesis is hardly new in the interpretation of neurological and psychiatric conditions, since an increased excitation:inhibition ratio has also been hypothesized to be a critical factor in the aetiology of anxiety (Mohler, 2006), schizophrenic psychoses (Kehrer et al. 2008), epilepsy (Fritschy, 2008; Trevino et al. 2011), insomnia (Mohler, 2006), depression (Sanacora et al. 2004) and catatonia (Daniels, 2009). The differences in the clinical manifestation of these disorders might originate in the timing (prenatal, early post-natal, or adult) and duration (acute vs. prolonged) of the biological insult, as well as in genetic predisposition. The pervasive nature of ASD symptoms might be rooted in an early peri-natal hyperexcitability at a critical time for storing and solidification of the complex set of social and communication rules implemented by the developing neocortex. In contrast, a recent optogenetics study (Yizhar et al. 2011) suggests that an imbalance between inhibition and excitation in favour of the latter can acutely impair social behaviour, independent of long-term circuit changes. Regardless of whether long- or short-term changes are necessary to produce autistic symptoms, our data corroborate the hypothesis that the increase in synaptic excitatory:inhibitory balance is a hallmark of ASD.
A lack of behavioural flexibility often observed in ASD patients might be, at least in part, secondary to a deficient control of GABAergic transmission by the modulatory systems, including the noradrenergic and the cholinergic systems (Salgado et al. 2007, 2011). The improvement in impulsivity, inattention, hyperactivity and aggressiveness symptoms, following administration of the α2 adrenergic agonist clonidine in a cohort of ASD children (Ming et al. 2008) would support this interpretation. The integrity of the GABAergic interneuronal network is essential in the generation of γ-oscillations, coordinating local with distant neuronal processing.
A deficit in GABAergic transmission implemented by local interneuronal network might then explain the abnormal γ-rhythms monitored in autistic subjects (Orekhova et al. 2007). It has been recently proposed that the failure to synchronize biological and circadian rhythms in new-borns might be a causal factor for ASD (Bourgeron, 2007). Consistent with this hypothesis we speculate that an early impairment of the GABAergic system might alter the extent and functional properties of local cortical excitatory circuits leading in turn to an enhancement of locally-controlled tasks but impaired higher-level processing. A corollary of this hypothesis is that treatment with allosteric modulators of synaptic GABAARs, or other drugs restoring the excitation:inhibition ratio to physiological levels might override the deleterious effects of the synaptic unbalance, as long as treatment occurs before the stabilization of the glutamatergic circuits for communication and social interactions.
Supplementary material
Supplementary material accompanies this paper on the Journal's website.
Supplementary information supplied by authors.
Acknowledgements
We thank Dr Crystal Engineer for advice during the execution of the project and for critical review of the manuscript. This work was funded with Research Development funds from the School of Behavioural and Brain Sciences to M.A. and with the National Institutes of Health R01DC010433 grant to M.P.K.
Statement of Interest
None.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}